La vida funciona gracias a moléculas diminutas que no vemos, pero que trabajan constantemente dentro de las células. Algunas transportan sustancias, otras aceleran reacciones químicas, otras envían señales y otras ayudan a construir tejidos. Entre todas ellas, las proteínas ocupan un lugar central.
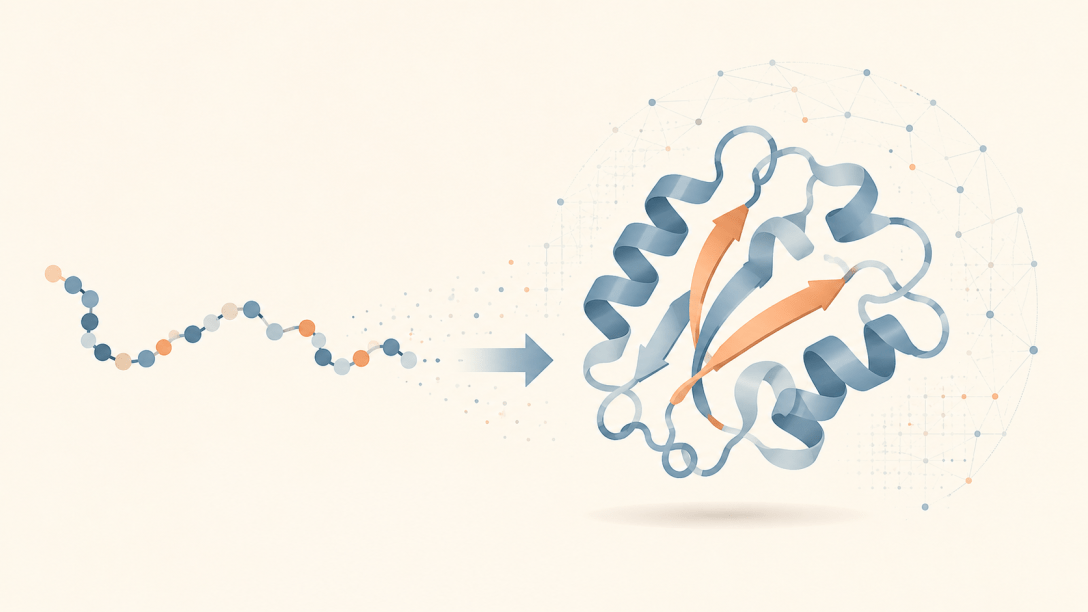
Una proteína es una molécula formada por una cadena de piezas más pequeñas llamadas aminoácidos. Podemos imaginar los aminoácidos como las letras de un alfabeto químico. Al combinarse en distinto orden, forman cadenas diferentes. Pero en biología no basta con conocer la secuencia de esas “letras”. Lo decisivo es la forma que adopta la cadena cuando se pliega en el espacio.
Esa forma tridimensional determina qué puede hacer una proteína. Si encaja con otras moléculas, si participa en una reacción, si transporta una sustancia o si está relacionada con una enfermedad depende, en gran parte, de su estructura. Por eso, durante décadas, una de las grandes preguntas de la biología fue: si conocemos la secuencia de aminoácidos de una proteína, ¿podemos predecir su forma?
El problema del plegamiento
El reto era mucho más difícil de lo que parece. Una cadena de aminoácidos puede plegarse de muchas maneras posibles, pero en la naturaleza acaba adoptando una estructura concreta. Conocer esa estructura era fundamental para entender su función, pero obtenerla mediante técnicas experimentales podía ser lento, caro y complejo.
El problema, por tanto, consistía en pasar de una información lineal —la secuencia de aminoácidos— a una estructura tridimensional. Era como intentar deducir la forma final de una figura compleja sabiendo únicamente el orden de sus piezas.
Durante años, este problema fue uno de los grandes desafíos de la biología estructural. No porque faltara interés, sino porque la relación entre secuencia y forma era extraordinariamente compleja. Había demasiadas interacciones, demasiadas posibilidades y demasiados detalles físicos para resolverlo con reglas simples.
AlphaFold como modelo predictivo
AlphaFold, desarrollado por DeepMind, abordó este reto como un problema de predicción cuantitativa. El modelo aprendió a partir de proteínas cuya secuencia y estructura ya se conocían experimentalmente. A partir de esos ejemplos, fue capaz de detectar patrones entre el orden de los aminoácidos y la forma final de la proteína.
En el mapa de métodos de previsión, AlphaFold se sitúa claramente en la familia del machine learning supervisado, y más concretamente en el aprendizaje profundo. No es una regresión sencilla ni una serie temporal. Es un modelo que aprende de muchos casos conocidos para predecir una salida compleja: una estructura 3D.
La entrada del modelo es la secuencia de aminoácidos. La salida no es un único número, sino una geometría: posiciones, distancias y relaciones espaciales entre las partes de la proteína. Por eso AlphaFold es una máquina de predicción especialmente sofisticada. No predice una cifra, sino una forma.
El salto científico
El gran impacto de AlphaFold fue demostrar que este tipo de predicción podía alcanzar una precisión muy alta. En la competición CASP14, una evaluación internacional de métodos de predicción de estructuras proteicas, AlphaFold obtuvo resultados muy superiores a los sistemas anteriores. Sus predicciones se acercaban, en muchos casos, a las estructuras observadas experimentalmente.
Este avance cambió la escala del problema. Lo que antes podía requerir meses de trabajo experimental podía tener ahora una primera aproximación computacional mucho más rápida. Además, la base de datos de AlphaFold puso a disposición de la comunidad científica millones de predicciones de estructuras proteicas.
Un ejemplo reciente aparece en la investigación contra la malaria. Algunos equipos han utilizado AlphaFold para comprender mejor la estructura de proteínas del parásito difíciles de estudiar experimentalmente, como Pfs48/45, una proteína relevante para bloquear la transmisión de la enfermedad. En este caso, la predicción no sustituyó al laboratorio, pero ayudó a orientar mejor qué partes de la proteína podían ser interesantes para diseñar futuras vacunas o anticuerpos.
Esto no eliminó la necesidad de la experimentación. Pero sí cambió el punto de partida. Los investigadores podían utilizar las predicciones de AlphaFold como hipótesis iniciales para estudiar enfermedades, diseñar proteínas, explorar nuevas moléculas o comprender mejor procesos biológicos.
El Nobel y una nueva forma de hacer ciencia
En 2024, el Premio Nobel de Química reconoció este cambio. David Baker fue premiado por el diseño computacional de proteínas, mientras que Demis Hassabis y John Jumper recibieron el reconocimiento por la predicción de estructuras proteicas mediante AlphaFold.
El Nobel no solo reconocía una herramienta concreta, sino una nueva manera de hacer ciencia. AlphaFold mostró que los modelos predictivos pueden ser algo más que instrumentos auxiliares. Pueden convertirse en una forma de descubrimiento.
La predicción, en este caso, no consistía en anticipar el futuro, sino en revelar una estructura oculta. La proteína ya tenía una forma posible, pero era difícil observarla. AlphaFold permitió estimarla a partir de datos y aprendizaje automático.
Conclusión: predecir para ver mejor
AlphaFold nos obliga a ampliar la idea de predicción. A veces predecir no significa decir qué ocurrirá mañana, sino estimar algo que existe pero no podemos ver fácilmente.
Esa es una de las grandes promesas de la inteligencia artificial aplicada a la ciencia: ayudarnos a descubrir patrones demasiado complejos para la intuición humana. No sustituye la experimentación ni elimina la necesidad de interpretar los resultados, pero puede acelerar enormemente el camino hacia nuevas preguntas.
AlphaFold no solo predijo proteínas. Nos enseñó que, cuando los datos y los modelos se combinan bien, predecir también puede ser una forma de mirar más profundamente la realidad.
